First of all, 50% of the CF population is homozygous (2 copies) for DF508, and 90% have a single copy. Researchers believe that approximately 30% of CFTR function is needed to get the type of clinical benefits seen with Kalydeco (big drops in sweat chloride paired with large gains in FEV1). This can be achieved by treating only 1 mutation, so this DF508 research affects almost everyone with CF. As I outlined in the blog entry “Mutation Matters,” http://luckycfmom.blogspot.com/2011/11/mutation-matters.html
cystic fibrosis mutations are placed into different classes, depending on HOW the CFTR protein is dysfunctional in the cell. Researchers are looking at two major concepts when classifying:
cystic fibrosis mutations are placed into different classes, depending on HOW the CFTR protein is dysfunctional in the cell. Researchers are looking at two major concepts when classifying:
I. How much CFTR matures properly and reaches the cell membrane
II. How much of that CFTR on the membrane actually opens and functions properly to transport Chloride.
For patients with G551D, the protein matures and reaches the appropriate place in the cell, but simply does not open enough for effective Chloride transport. Kalydeco makes that channel open, and restores Chloride function similar to what carriers of the CF gene exhibit—around 50%. Sweat chloride levels dropped 47-48mmol on average. Brady’s baseline sweat test was 105. If his score drops the average amount (we will find out next Wednesday!), he will go to a 58, which is considered the inconclusive gray area for diagnosis of CF.
The dysfunction in the DF508 mutation is much more complex and this is the detail I’m really trying to get to in this entry. For reference, we need a model of the CFTR protein.
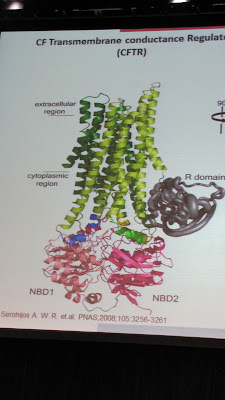
Think of it as a piece of origami. Every single fold must be made correctly, and in the right order, or it will get crumpled up and thrown in the trash. DF508’s first problem is that it gets misfolded in nucleotide binding domain 1 (NBD1). Technically, a base pair is missing from the genetic sequence, which is why it is sometimes called a “deletion” mutation. Anyway, correctors like VX-809 work to chemically fill that deletion and restore the folding mechanism. I am hopeful because the amount of protein that they could restore through combination treatment was dose dependent (more VX-770, more CFTR), and also, that they never reached a ceiling or peak on clinical improvement in the short amount of time that patients were on the combo (only 1 week), so they are truly unsure what pumped up dosages might achieve. VX-809 degrades the action of VX-770, so when the two drugs are combined, they found that they needed to increase the normal dosage of VX-770 to get the best effects. I am curious and hopeful that a higher dosage combination might be able to reach that critical threshold of benefit and proceed to phase 3 clinical trials. In vitro (lab), they were able to achieve a 35% restoration with the combo, so it has a real chance. The ongoing clinical trial will determine if this same restoration can be reached when actually administered to live people. If this combo were to succeed and reach the market, it is estimated for 2016.

Think of it as a piece of origami. Every single fold must be made correctly, and in the right order, or it will get crumpled up and thrown in the trash. DF508’s first problem is that it gets misfolded in nucleotide binding domain 1 (NBD1). Technically, a base pair is missing from the genetic sequence, which is why it is sometimes called a “deletion” mutation. Anyway, correctors like VX-809 work to chemically fill that deletion and restore the folding mechanism. I am hopeful because the amount of protein that they could restore through combination treatment was dose dependent (more VX-770, more CFTR), and also, that they never reached a ceiling or peak on clinical improvement in the short amount of time that patients were on the combo (only 1 week), so they are truly unsure what pumped up dosages might achieve. VX-809 degrades the action of VX-770, so when the two drugs are combined, they found that they needed to increase the normal dosage of VX-770 to get the best effects. I am curious and hopeful that a higher dosage combination might be able to reach that critical threshold of benefit and proceed to phase 3 clinical trials. In vitro (lab), they were able to achieve a 35% restoration with the combo, so it has a real chance. The ongoing clinical trial will determine if this same restoration can be reached when actually administered to live people. If this combo were to succeed and reach the market, it is estimated for 2016.
If you have been following the Vertex clinical trials, that last paragraph may not have been newsworthy. There is truly no way of knowing how that combo will perform when dosages are optimized until the data is released. I have been seeing a lot of questions and confusion about “second generation” correctors. This projected next step in treatment is the result of an increasing body of knowledge researchers are accumulating about DF508. It was recently discovered that DF508 has a second major obstacle to cell maturation (messing up the origami swan). The CFTR protein has 2 major parts (nucleotide binding domains or NBD) that must interact with one another for perfect protein assembly. The second major problem facing the DF508 mutation, is instability at the CL4 interface. This interface is the place where NBD1 and NBD2 come together.

This interface problem can also be improved with a different type of corrector compound, designed to stabilize this site. They found that when two different corrector compounds with different actions(one to fix misfolding, one to stabilize interface) were combined, they worked synergistically-- in other words--the combined result is greater than the sum of the two individually. This means you might get 20% restored with one corrector alone, and 20% out of the other corrector alone, but when combined…20% + 20% = 70% or more CFTR restoration!

VX-770 will absolutely be a part of this combination, because even when DF508 proteins fold and assemble properly—they also exhibit a gating defect if they manage to make it to the cell membrane. This is where VX-770 comes in. VX-770 works to open ANY CFTR that makes it to the cell membrane, thereby overcoming the gating defect. DF508 is also suspected to suffer increased cell turnover/premature cell death (defect similar to class 6 mutations) at the cell surface, further reducing the ability to transport Chloride. So let’s review all the different issues that must be overcome to restore function of the DF508 mutation:

This interface problem can also be improved with a different type of corrector compound, designed to stabilize this site. They found that when two different corrector compounds with different actions(one to fix misfolding, one to stabilize interface) were combined, they worked synergistically-- in other words--the combined result is greater than the sum of the two individually. This means you might get 20% restored with one corrector alone, and 20% out of the other corrector alone, but when combined…20% + 20% = 70% or more CFTR restoration!

VX-770 will absolutely be a part of this combination, because even when DF508 proteins fold and assemble properly—they also exhibit a gating defect if they manage to make it to the cell membrane. This is where VX-770 comes in. VX-770 works to open ANY CFTR that makes it to the cell membrane, thereby overcoming the gating defect. DF508 is also suspected to suffer increased cell turnover/premature cell death (defect similar to class 6 mutations) at the cell surface, further reducing the ability to transport Chloride. So let’s review all the different issues that must be overcome to restore function of the DF508 mutation:
1) Initial NBD1 misfolding due to deletion of base pair in genetic sequence—needs corrector compound like VX-809 or something similar to restore proper folding.
2) Instability at the CL4 interface—would require an additional corrector molecule with a different mechanism of action to help restore stability at this assembly site.
3) Proteins that do successfully mature and reach the cell surface, also exhibit a gating defect similar to class 3 mutations. VX-770 will be needed to ensure the CFTR channels that make it to the surface actually open and restore Chloride transport.
4) Increased cell turnover at surface means that it is even more important to include a potentiator like VX-770 to keep the channels open as much as possible to achieve best results.
In conclusion, I want to say that by including information about the “second generation” correctors, I am in no way implying that the first generation correctors won’t succeed to market and make a huge impact on the CF community. There is a real chance for a first generation treatment to be successful. BUT, no matter what, the second generation combinations of 2 correctors plus potentiator have shown in the lab to be incredibly effective and those are projected for a 2020 market date. Restoring even a modest amount of CFTR can slow the progression of CF, so a first generation could at the very least, buy some time. At the very best, it may work as well as VX-770 works for G551D. I can’t wait to see. And I know I have said this before, but I truly believe they have solved the puzzle for DF508, that now it is a matter of time and money to see these molecules through to the market. I know many people have wondered why the G551D mutation was targeted first, rather than DF508, which would affect a much larger group of people. The fact is that G551D has one simple dysfunction to correct--open the gate. In addition, VX-770 was one of the first molecules discovered to have activity with the CFTR protein. With DF508, restoring Chloride transport is much more challenging and complex. You have to first build the gate before you have any hope of opening it. Here is a very simple white board explanation of the action of VX-770 and VX-809 that I made about a year ago.
One final note for Class 1 or stop mutations. I sense the frustration with the lack of data on the final Ataluren trial. 10% of CFers have a mutation in this class and are curious to see whether this is a viable option or not. I don’t have any inside data on the effectiveness of Ataluren alone, but one presenter at the NACFC talked at length about the lab data that VX-770 might be combined with a drug to promote translational read-through of the protein (what Ataluren is supposed to do) to increase Chloride transport activity. I guess I wanted to include this data because even if Ataluren alone doesn’t prove to be the magic bullet for nonsense mutations, that doesn’t necessarily mean back to the drawing board. VX-770 may be able to be combined with a drug like Ataluren to increase the CFTR rescue for this mutation class. Because VX-770 is already FDA approved, it would shave years off the clinical trial process, so that is great news.
For Reference:
You can check out a lot of this information for yourself in the first Plenary session at the NACFC last fall: http://www.cff.org/research/NACFC/2011NACFC/
Additional information was taken from my notes from:
"Combination of Correctors & Potentiators for ΔF508"
Track : Symposium Session III Date: Saturday, November 05, 2011 NACFCSpeaker Steven Rowe from University of Alabama at Birmingham and John Boyle from John's Hopkins
Abstract 4. The CFTR Folding Pathway: Steps Altered by ΔF508 & Implications for the Discovery & Development of CF Therapeutics
Track : Workshop Session I Date: Thursday, November 03, 2011 NACFCSPEAKER :
Juan Luis Mendoza from UT Southwestern Medical Center at Dallas
Abstract 6. Methods to Establish Mechanism of Corrector Action
Track : Workshop Session II Date: Friday, November 04, 2011 NACFC
SPEAKER : was supposed to be Philip Thomas, Ph.D., UT Southwestern Medical Ctr., but was replaced by his colleage and formerly mentioned Juan Luis Mendoza.
Quick Kalydeco Update:
Brady is now 2 weeks deep in his Kalydeco treatment. He seems like he feels fantastic. Tons of energy, and sleeping better at night. We have reduced his steroid sinus rinses from 3X/day down to once a day and he is maintaining clear sinuses. He has gained a little over 1 lb. since he started the pill. I have only slightly decreased his enzyme dosage(I've been giving him about 2.5 rather than 3 Zenpep per meal). We go in to repeat the sweat test next Wednesday. We will repeat bloodwork and see his CF doc again on March 8th (1 month from beginning treatment). So far I say two thumbs WAY UP for Kalydeco!
Oh man fantastic post. Our 8-month-old daughter has F508del and we're anxiously awaiting the news on the Vertex trials.
ReplyDeleteThis is a great post. I shared it with most of my family - it explains the "science" of the answer to all our prayers very well.
ReplyDeleteThanks so much for this post, fantastic, and so clearly explained. I have been waiting years for someone to explain all the details and differences in strains and its problems. I have a double copy of D-F508. I´m now 30 years old, married and most amazing of all managed to finally have my miracle baby Kyra Rose. I wouldn´t change the life I have now with CF for a life without Cf and no baby Kyra and husband Jose. it´s a small price to pay for such a happy and amazing life.
ReplyDeleteyeezy boost 350 white
ReplyDeleteray ban sunglasses
instyler max 2
canada goose outlet
michael kors outlet
cheap michael kors handbags
michael kors handbags
tiffany and co
kobe 9
cheap ray ban sunglasses